
OakVar is a Python-based platform for analyzing genomic variants. One predominant purpose of OakVar is simple integration of genomic information with AI/ML frameworks. On this tutorial, I’d like to point out how simply such integration may be finished.
We’ll prepare classifiers which can predict the ancestry of an individual utilizing the particular person’s genotype information.
Let’s say we’re contemplating solely 5 variants, sampled from 5 folks, every of whom belongs to considered one of two populations, African and European. These 5 folks’s ancestry and variant presence are beneath.
Variants
Identify Ancestry A B C D E
Jack African o o x x x
Paul European x x o o o
James European x x x o o
John African o o o x x
Brian African o o x x x
We will think about the above as the next.
y X
African 1 1 0 0 0
European 0 0 1 1 1
European 0 0 0 1 1
African 1 1 1 0 0
African 1 1 0 0 0
Utilizing scikit-learn library of Python and the above information, a classifier to foretell ancestry based mostly on these 5 variants may be skilled as follows.
import numpy as np
from sklearn.naive_bayes import MultinomialNB
from sklearn.model_selection import train_test_splitX = np.array([
[1,1,0,0,0],
[0,0,1,1,1],
[0,0,0,1,1],
[1,1,1,0,0],
[1,1,0,0,0]
])
y = np.array([
"African",
"European",
"European",
"African",
"African"
])
clf = MultinomialNB()
X_train, X_test, y_train, y_test = train_test_split(X, y)
y_pred = clf.match(X_train, y_train).predict(X_test)
print("y_test:", y_test)
print("y_pred:", y_pred)
y_test: ['African' 'European']
y_pred: ['African' 'European']
This was a toy instance. The true information we are going to use is The 1000 Genomes Project, which has the identical form of ancestry and genotyping information of greater than 2,000 folks from world wide.
As a fast check, we are going to use the chromosome Y dataset of the 1000 Genomes Challenge, considered one of its smallest datasets. This dataset has the genotype and phenotype information of 1,233 folks.
There are three recordsdata to obtain. Please obtain them here, here, and here.
Afterwards, the next three recordsdata must be current in your working listing.
ALL.chrY.phase3_integrated_v1b.20130502.genotypes.vcf.gz
20130606_g1k.ped
20131219.populations.tsv
The .vcf.gz file is genotyping information. The .ped and .tsv recordsdata are ancestry information.
If you’re new to OakVar, please set up it as defined in the instruction. Briefly, open a command-line interface with a Python surroundings and run
pip set up oakvar
to put in OakVar and
ov system setup
to arrange OakVar in your system.
Please set up the next Python libraries.
pip set up scikit-learn
pip set up numpy
pip set up matplotlib
pip set up polars
I’ll use JupyterLab for interactive evaluation and visualization. If you happen to don’t have it but, please set up it in accordance with their instruction. Then, launch it with
jupyter-lab
On the JupyterLab, select a Python 3 pocket book. The codes from listed here are alleged to be entered on a JupyterLab pocket book.
Let’s load the chromosome Y information into an OakVar consequence database.
!ov run ALL.chrY.phase3_integrated_v1b.20130502.genotypes.vcf.gz
This can carry out primary annotation of the genotype information and write the consequence to ALL.chrY.phase3_integrated_v1b.20130502.genotypes.vcf.gz.sqlite.
If you’re , the evaluation consequence may be explored in a GUI with
!ov gui ALL.chrY.phase3_integrated_v2b.20130502.genotypes.vcf.gz.sqlite
See here for easy methods to use the GUI consequence viewer. Interrupt the kernel of JupyterLab to complete the viewer.
As proven to start with of this submit, we want X and y arrays to coach and check classifiers with scikit-learn. X will probably be a 2D array of variants. y will probably be a 1D array of ancestry.
OakVar gives a way with which we will assemble such X and y from genotyping information. Utilizing the annotation consequence within the earlier part,
import oakvar as ovsamples, uids, variants = ov.get_sample_uid_variant_arrays(
"ALL.chrY.phase3_integrated_v2b.20130502.genotypes.vcf.sqlite"
)
ov.get_sample_uid_variant_arrays returns three NumPy arrays. The primary one (samples) is a 1D array of pattern names.
array(['HG00096', 'HG00101', 'HG00103', ..., 'NA21130', 'NA21133',
'NA21135'], dtype='<U7')
The second (uids) is a 1D array of the variant UIDs.
array([ 1, 2, 3, ..., 61868, 61869, 61870], dtype=uint32)
The third one (variants) is a 2D array of the presence of variants.
array([[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
...,
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.]])
The connection of the three arrays are: samples describes the rows of variants with the title of the pattern of every row of variants, and uids describes the columns of variants with the variant UID of every column of variants.
We don’t have X and y but. We nonetheless want some transformation to get them.
The 1000 Genomes challenge information is organized in a number of ranges. The primary degree is samples, that are particular person individuals. Samples are grouped into populations, that are proven beneath.
Japanese in Tokyo, Japan
Punjabi in Lahore, Pakistan
British in England and Scotland
African Caribbean in Barbados
...
Populations are grouped into tremendous populations and there are 5 tremendous populations within the 1000 Genomes challenge information.
AFR = African
AMR = American
EAS = East Asian
EUR = European
SAS = South Asian
The chromosome Y information is just too small to foretell populations. Thus, we are going to purpose to foretell tremendous populations. The rows of variants array we retrieved within the earlier part correspond to samples. We’d like an array of tremendous populations which corresponds to samples array.
import polars as pldf_p = pl.read_csv(
"20131219.populations.tsv",
separator="t"
).choose(
pl.col("Inhabitants Code", "Tremendous Inhabitants")
).drop_nulls()
df_s = pl.read_csv(
"20130606_g1k.ped",
separator="t",
infer_schema_length=1000
).choose(
pl.col("Particular person ID", "Inhabitants")
).drop_nulls()
df_s = df_s.be part of(
df_p,
left_on="Inhabitants",
right_on="Inhabitants Code"
)
superpopulations = [
df_s["Super Population"][np.where(df_s["Individual ID"] == v)[0]][0]
for v in samples
]
superpopulations will probably be a 1D array which corresponds to the rows of variants. That is our y.
For X, variants has too many columns (61,870 columns), permitting over-training. We use principal element evaluation (PCA) to cut back its dimensionality to 2 columns.
from sklearn import decompositionpca = decomposition.PCA(n_components=2)
pca.match(variants)
X = pca.rework(variants)
We now have our X and y for coaching and testing ancestry classifiers.
y X
EUR -22.72643988 -22.48927295
EUR -8.68957706 13.9559764
EUR -22.78267228 -22.48553873
...
Let’s visualize this information.
mport matplotlib.pyplot as pltcolor_by_superpopulation = {
"AFR": "#ff0000",
"EAS": "#00ff00",
"EUR": "#0000ff",
"AMR": "#00ffff",
"SAS": "#ff00ff",
}
colours = [color_by_superpopulation[v] for v in superpopulations]
fig = plt.determine()
ax = fig.add_subplot()
ax.scatter(X[:, 0], X[:, 1], c=colours)
plt.present()
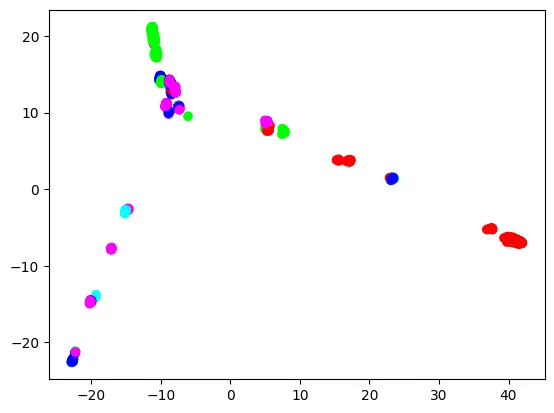
Every dot is a pattern, coloured by the pattern’s tremendous inhabitants. General, Europeans (blue), East Asians (inexperienced), and Africans (pink) make three extremities, and Individuals (cyan) and South Asians (purple) join these three extremities. Nonetheless, European dots should not clearly separated from different tremendous inhabitants dots. In the meantime, East Asian and African dots are extra clearly separated. That is comprehensible, as a result of the European tremendous inhabitants within the 1000 Genomes Challenge contains British, Spanish, Finnish, Italian, and folks of Northern and Western European ancestry, so it’s already fairly numerous. We’ll see if skilled classifiers will replicate this side.
The primary form of classifiers we are going to check is K-nearest neighbor (k-NN) classifier.
from sklearn.neighbors import KNeighborsClassifier
from sklearn.model_selection import train_test_splitclf = KNeighborsClassifier()
y = superpopulations
X_train, X_test, y_train, y_test = train_test_split(X, y)
y_pred = clf.match(X_train, y_train).predict(X_test)
This classifier’s precision, accuracy, and F1 rating are:
Precision F1 rating
AFR 0.924 0.912
AMR 0.548 0.430
EAS 0.962 0.971
EUR 0.532 0.596
SAS 0.838 0.844Accuracy: 0.773
Good efficiency with Africans and East Asians and decrease efficiency with Europeans agree with what we noticed within the PCA plot within the earlier part.
The following is Random Forest classifier.
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import train_test_splitclf = RandomForestClassifier(n_estimators=10)
y = superpopulations
X_train, X_test, y_train, y_test = train_test_split(X, y)
y_pred = clf.match(X_train, y_train).predict(X_test)
This classifier’s precision, accuracy, and F1 rating are:
Precision F1 rating
AFR 0.944 0.944
AMR 0.478 0.543
EAS 0.981 0.981
EUR 0.642 0.602
SAS 0.924 0.897Accuracy: 0.825
This one performs barely higher than the k-NN one, nevertheless it has the identical attribute that it performs higher with Africans and East Asians than with Europeans.
get_sample_uid_variant_arrays can be utilized to selectively retrieve samples and variants.
samples, uids, variants = get_sample_uid_variant_arrays(
"ALL.chrY.phase3_integrated_v2b.20130502.genotypes.vcf.sqlite",
variant_criteria="gnomad3__af > 0.01"
)
will return variants the gnomAD allele frequency of which is larger than 1%, if the enter file was annotated with gnomad3 OakVar module as in
ov run ALL.chrY.phase3_integrated_v2b.20130502.genotypes.vcf.sqlite -a gnomad3
samples and uids will probably be adjusted accordingly as properly. Any OakVar module can be utilized for such filtration.
This was a fast demonstration of OakVar’s skill to interface genomic information and scikit-learn, a machine studying framework. Please keep tuned for future publications for extra examples.